商品名称:QIAseq 16S/ITS Smart Control (10)
QIAseq 16S/ITS Panels are the Sample to Insight solution for robust profiling of bacterial and fungal communities. The panels have been developed for sequencing 16S rRNA and ITS regions on Illumina platforms. Unlike other solutions, QIAseq 16S/ITS Panels use "phased primers" to increase the quality of reads and base calling, and eliminate the need for PhiX spike-in. In addition, QIAseq 16S/ITS Panels incorporate low-bioburden reagents to decrease background contamination, interrogate all 16S rRNA variable regions and allow low DNA input, enabling the analysis of low-biomass samples.
The QIAseq 16S/ITS Region Panels, when combined with any of the QIAseq 16S/ITS Index Kits, include all required reagents to produce libraries from extracted DNA. Region panels can be configured to target 1, 2 or 3 16S variable regions and/or ITS. To configure your QIAseq 16S/ITS Region Panel, choose a product on the "Ordering Information" tab and click "Configure".
High quality of reads and bases
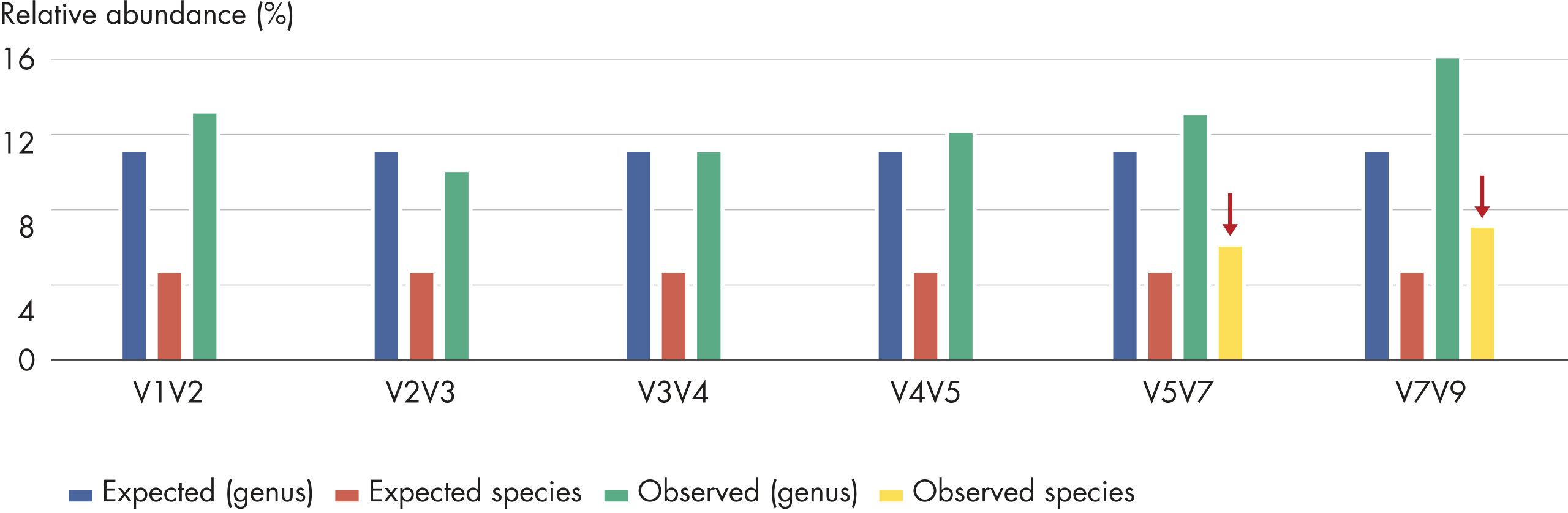
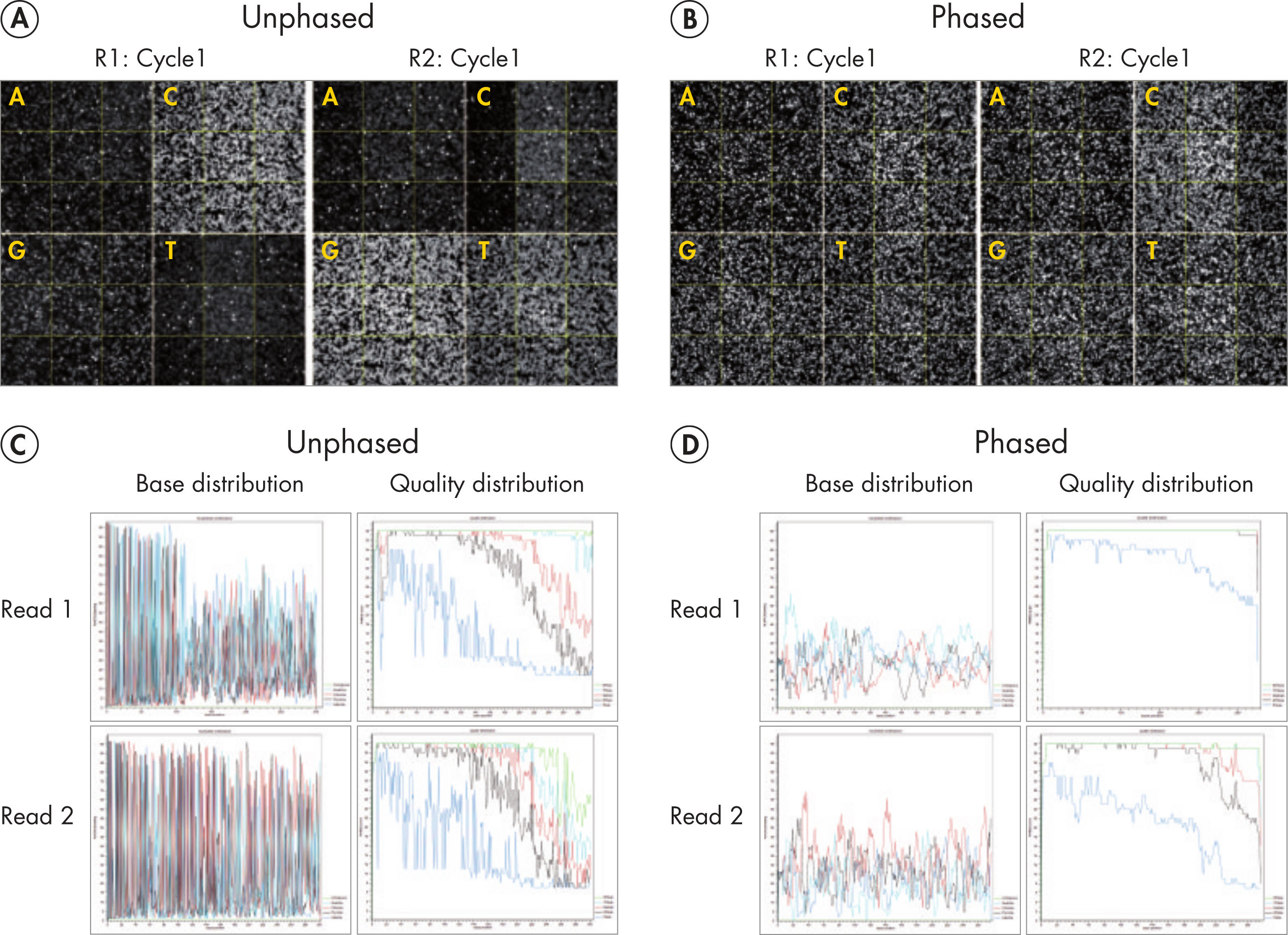
Sequencing single amplicon libraries often yields results of low quality due to the reduced diversity in base composition in the primer regions (figure QIAseq 16S/ITS Panels employ phased primers to increases base diversity and quality scores, A and C). To overcome this issue, QIAseq 16S/ITS Panels use a "phased primer" approach (figure Structure of phased primers used in QIAseq 16S/ITS Panels), which incorporates 0–11 additional bases to the 5’-end of the 16S rRNA or ITS primer. The use of "phased primers" results in a shift in nucleotide balance and increases base diversity, leading to an increase in quality scores (figure QIAseq 16S/ITS Panels employ phased primers to increases base diversity and quality scores, B and D).
Efficient use of flow cell throughput
Because of the "phased primer" approach, libraries produced with QIAseq 16S/ITS Panels are sufficiently complex to eliminate the need for PhiX spike-in, thereby enabling the efficient utilization of flow cell throughput.
Low background noise
Bacterial DNA is present in every corner of our daily lives, resulting in an increased risk of contamination during handling and processing of biological samples. Furthermore, manufacturing and processing of enzymes and reagents can introduce exogenous bacterial DNA to the samples being studied. This contamination background decreases the robustness of bacterial profiling. QIAseq 16S/ITS Panels use low-bioburden reagents, which show very low levels of exogenous bacterial contamination in NTC runs (figure The QIAseq 16S/ITS Panels have very low levels of background contamination due to the use of reagents with low bioburden).
Low DNA input
QIAseq 16S/ITS Panels can be used with a bacterial DNA input ranging from 1 pg to 1 ng, allowing users to profile bacterial communities in samples with low biomass (figure The QIAseq 16S/ITS Panels can be used with as little as 1 pg of input DNA). For profiling fungal communities/species, QIAseq 16S/ITS Panels can detect 0.01 ng of fungal DNA in a background of 1 ng of E. coli DNA.
Multiplexing
Each panel can be used to multiplex up to 96 samples on an Illumina MiSeq run using the V3 sequencing chemistry at 2x300. The appropriate index kit is necessary to multiplex the required number of samples.
Data analysis
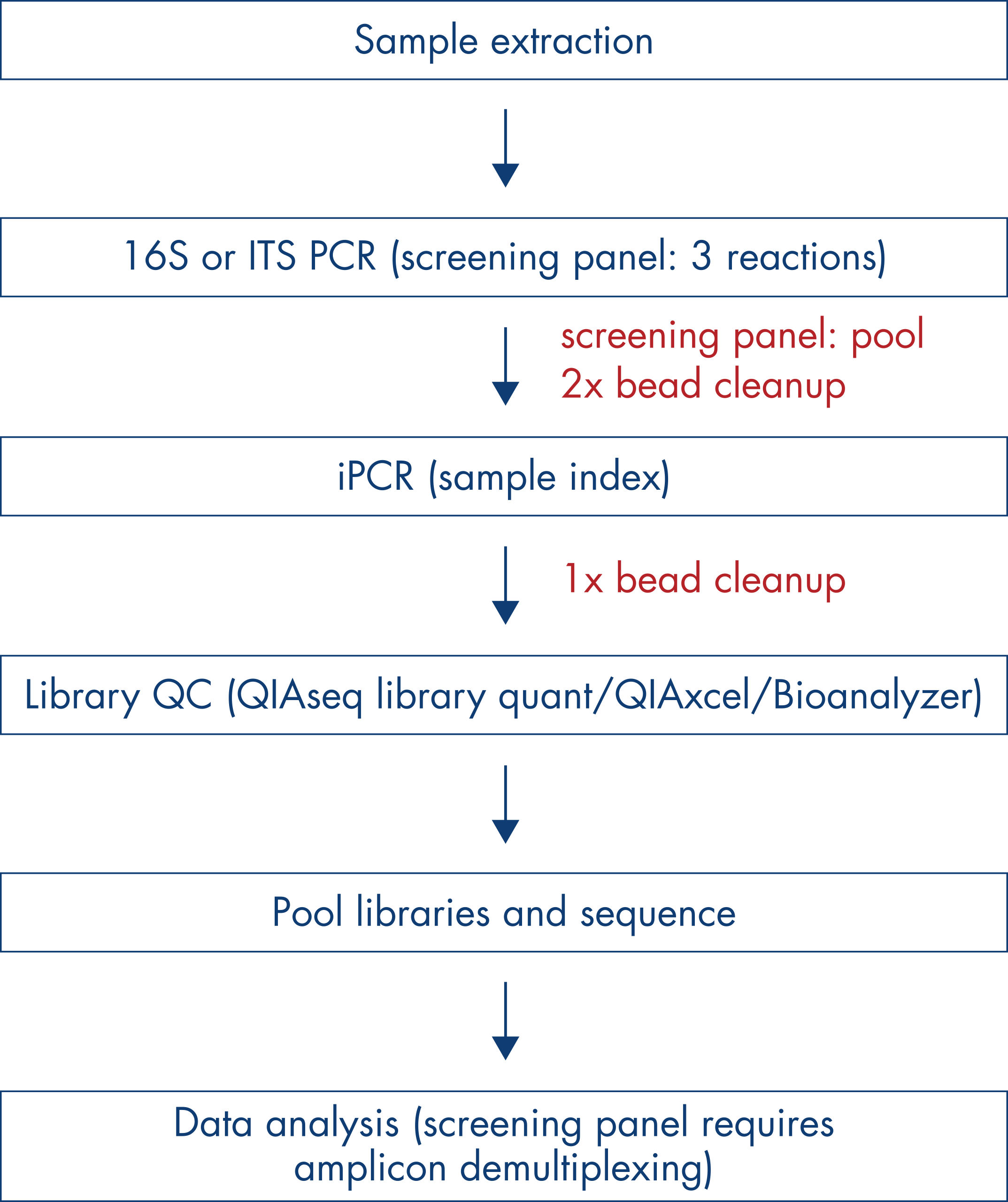
After sequencing, data is analyzed using QIAGEN Bioinformatics CLC Genomic Workbench and Microbial Genomics Pro Suite Module. A custom workflow, which can be further edited as needed, is available for automating FASTQ file import, sample library demultiplexing, quality controlled filtering and trimming, OUT clustering and secondary bioinformatics analysis. QIAGEN Bioinformatics CLC Genomics Workbench with the Microbial Genomics Pro Suite Module enables researchers to produce standardized reports on sample library quality metrics and organism abundance tables, providing numerous options for downstream interactive analysis of microbiome profiles and reporting on experimental results. The Microbial Genomics Pro Suite Module also includes tools for calculating diversity metrics and for comparing the microbial profiles of different samples. CLC Genomics Workbench produces publication quality figures in formats that are readily useable for researchers to present their results.
 Structure of phased primers used in QIAseq 16S/ITS Panels.
Structure of phased primers used in QIAseq 16S/ITS Panels. The QIAseq 16S/ITS Panels have very low levels of background contamination due to the use of reagents with low bioburden.
The QIAseq 16S/ITS Panels have very low levels of background contamination due to the use of reagents with low bioburden.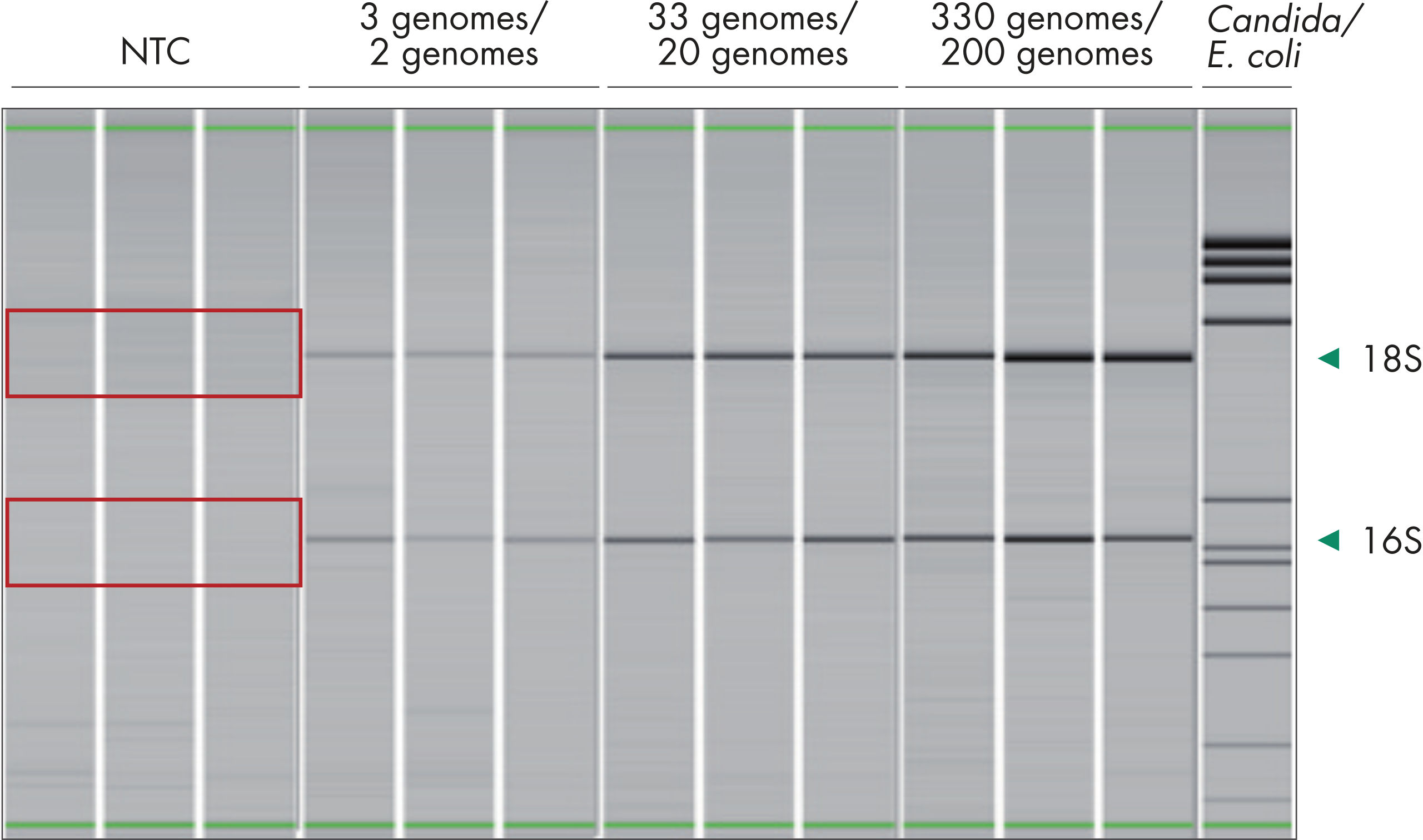 The QIAseq 16S/ITS Panels can be used with as little as 1 pg of input DNA.
The QIAseq 16S/ITS Panels can be used with as little as 1 pg of input DNA.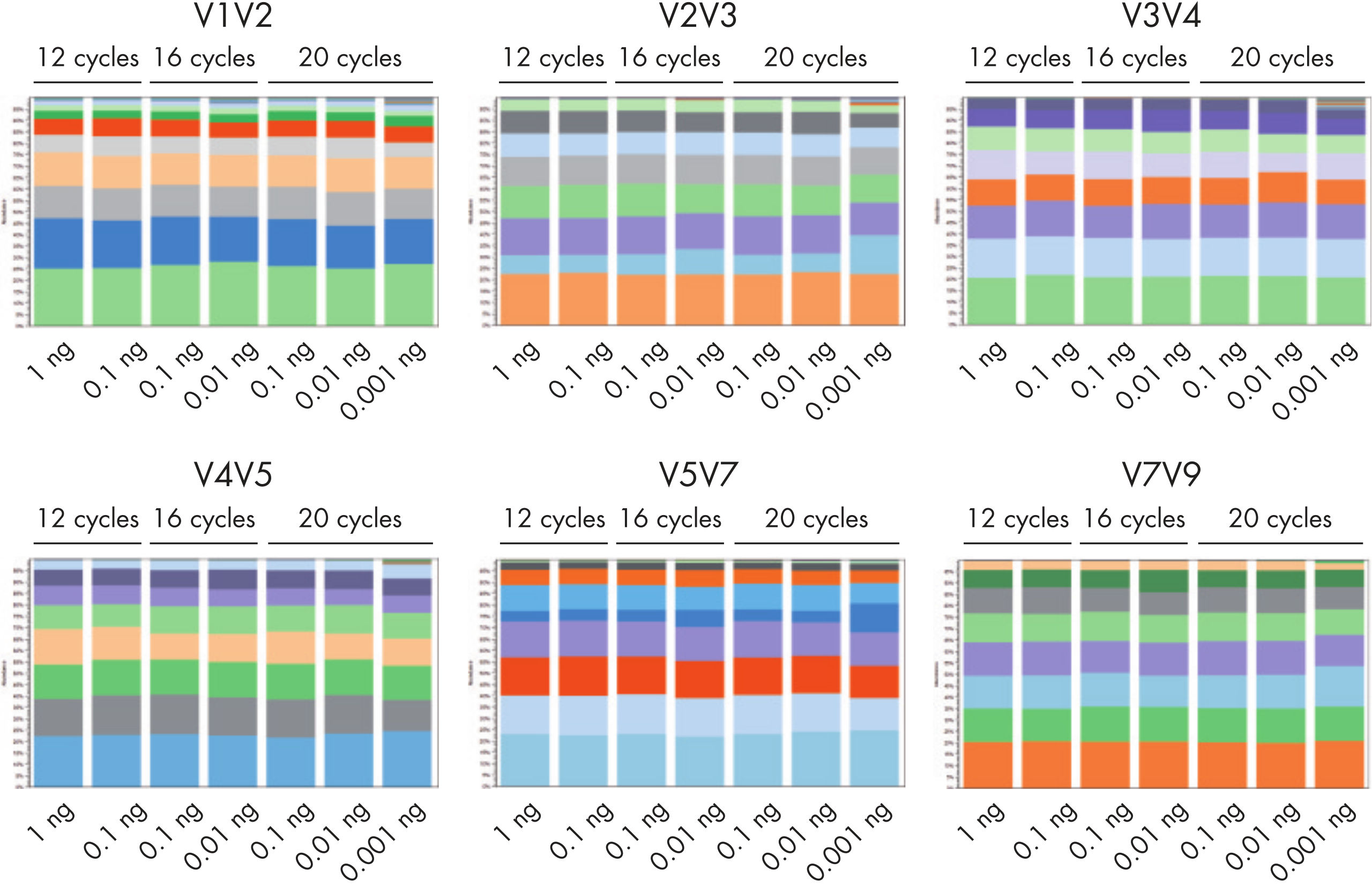
Introduction
The most common method used for profiling microbial communities is sequencing the 16S ribosomal RNA (rRNA) gene and Internal Transcribed Spacer (ITS) regions for bacteria and fungi, respectively. Because of the universal distribution and conserved nature of the 16S rRNA and ITS genes, they are well-established genetic markers used for bacterial and fungal identification and classification.
16S and ITS genes
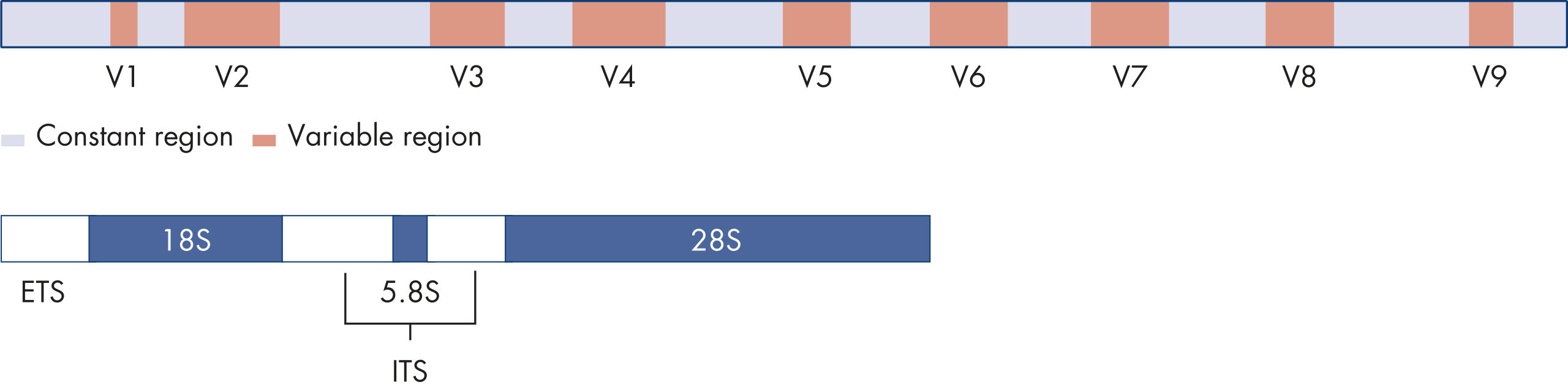
The 16S rRNA gene consists of both highly conserved and hypervariable regions (figure Structure of the bacterial 16S rRNA gene [top] and the fungal ITS region [bottom]). The conserved regions serve as primer binding sites for PCR amplification of the variable regions and the variable regions contain sequences that can be used for bacterial identification and classification.
The ITS region is situated between the small and large ribosomal RNA (rRNA) subunits. In eukaryotes, there are two ITS regions: ITS1 is located between the 18S rRNA and 5.8S rRNA genes, while ITS2 is located between the 5.8S rRNA and 28S rRNA genes (figure Structure of the bacterial 16S rRNA gene [top] and the fungal ITS region [bottom]). ITS regions have been shown to have the highest probability of successful identification for the broadest range of fungi.
Benefits of NGS for 16S rRNA gene and ITS sequencing
NGS provides a culture-free method for analyzing the microbial community within a biological sample. As the throughput of modern NGS systems has increased, it is not uncommon for libraries from multiple biological samples to be multiplexed on the same sequencing run, which has in turn enabled microbiology researchers to cost-effectively analyze hundreds of microbial communities in parallel. Determining the sequence of universal marker genes such as the 16S rRNA gene and ITS with NGS is a powerful method for surveying microbial communities as it can be used to:
Current challenges with 16S rRNA and ITS sequencing
Difficulties with NGS of 16S rRNA genes and ITS regions include:
Smart Control
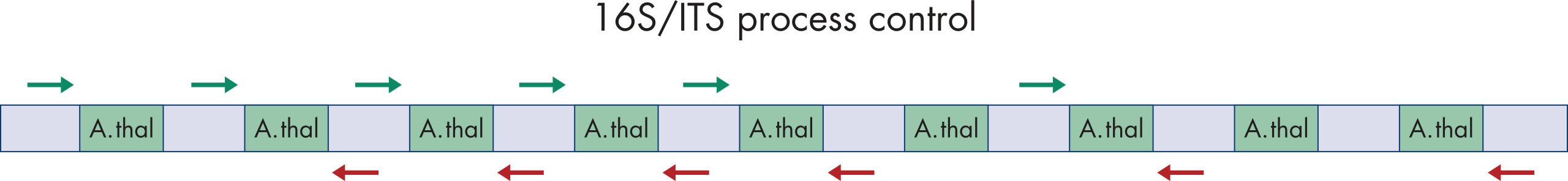
QIAseq 16S/ITS Smart Control is a synthetic DNA that can be used as a positive control for library construction steps as it contains the primer binding sites from Escherichia coli. The hypervariable 16S rRNA region between the primer binding sequences is replaced with artificial sequences originating from Arabidopsis thaliana (figure Structure of the QIAseq 16S/ITS Smart Control DNA). The artificial sequences cannot be classified as bacterial or fungal, therefore any sequences that are classified are due to environmental contamination introduced during library construction.
Copyright © 2016-2025 汇佰生物科技(深圳)有限公司 版权所有 保留一切权利 备案号 粤ICP备2022123097号-1  粤公网安备 44031102000815号
粤公网安备 44031102000815号